
Today We’re gonna be talking about the different regulatory Pathways that the FDA uses to control the approval of medical devices usually before you can market your medical device it has to be FDA approved or FDA cleared in most cases before you can even sell so as you may imagine there is a very wide range of different medical devices that there can meet and this is why the FDA has several regulatory pathways that you can follow depending on your device so, for example, the FDA is not gonna review a toothbrush same way that I review a pacemaker that is implanted near your heart you know this is why it’s very important to first know the classification of your device FDA classifies medical devices in three main classes which are class 1,2&3 as the class number increases so does the risk associated with it there’s actually a number of devices that are exempt from having to get a premarket notification or giving it to the FDA.

Three main classes of FDA classify medicals device:
- Least risk. Least Regulated: general controls
- Moderate risk and regulation: general controls and Special Controls
- Most risk and regulatory control: general controls and Premarket Approval (PMA)
General Controls:
General controls are described in the following sections of the FD&C Act:
- 501: Adulterated devices
- 502: Misbranded devices
- 510: Registration of producers of devices
- Establishment registration and device listing
- Premarket Notification (510k)
- Reprocessed single-use devices
- 516: Banned devices
- 518: Notifications and other remedies

- Notification
- Repair
- Replacement
- Refund
- Reimbursement
- Mandatory recall
- 519: Records and reports on devices
- Adverse event report
- Device tracking
- The unique device identification system
- Reports of removals and corrections
- 520: General provisions respecting the control of devices intended for human use
- Custom device
- Restricted device
- Good manufacturing practice requirements
- Exemptions for devices for investigational use
- Transitional provisions for devices considered as new drugs
- Humanitarian device exemption
general controls and Special Controls:
Special controls are usually device-specific and include:
- Performance standards
- Postmarked surveillance
- Patient registries
- Special labeling requirements
- Premarket data requirements
- Guidelines
Premarket Approval (PMA)
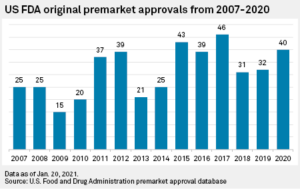
Under federal law, class III devices are subject to the approval of a Premarket Approval Application (PMA).
Devices that are not within a type marketed beforehand the date of the Medical Device Amendments of 1976 – referred to as reamendments devices – are confidential into class III automatically under federal law. In addition, the FDA classifies into class III devices intended to be used in backup or sustaining human life or averting impairment of human health, or that may present a potential irrational risk of illness or injury for which general controls and special controls are insufficient to deliver reasonable assurance of the safety and efficiency of a device, or for which there is inadequate material to make such a determination.